研究支援
疾患モデル研究センターの使命の一つとして「医学研究の発展のために新しい研究技術を取り入れて、より高度な研究に応じた支援体制を整える」ことを掲げております。
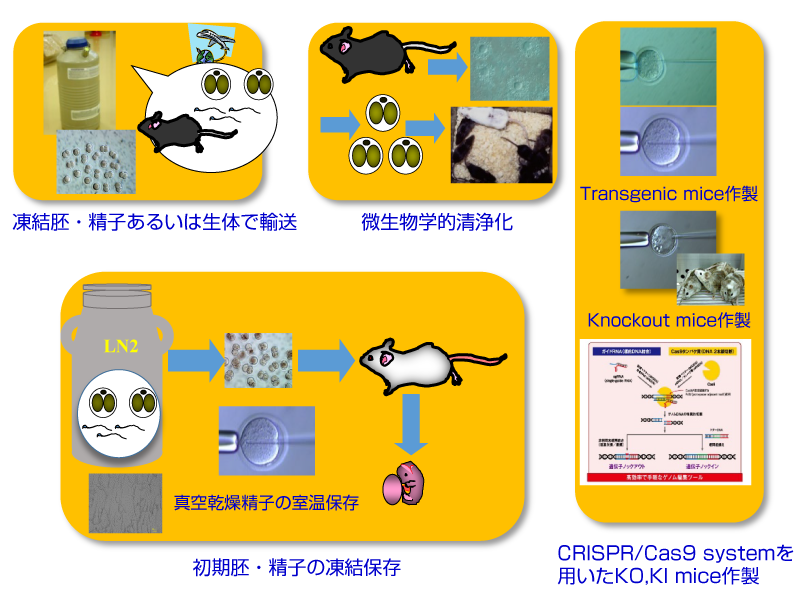
動物実験に使用する実験動物を外部研究機関から導入する場合、微生物感染が危惧されますので、指定された動物供給業者以外の大学等研究機関からの動物の導入時は、必ず、クリーニング(清浄化)を行います。クリーニングした動物、特に遺伝子改変マウスなどは、当センターの動物実験施設内で繁殖・維持しながら、実験に使用することになります。しかし、繁殖・維持している過程で突然変異などが起こり、その系統(ライン)の特性(表現型)が失われることがあります。そこで、導入後、できるだけ早く体外受精などで初期胚(2-細胞期胚)を得て、凍結保存しておけば、系統の表現型が変わった時点で、凍結胚の一部を融解して個体化すれば、系統の特性(表現型)を復元することができます。また、余剰な動物や実験終了後の動物は、系統保存(遺伝資源の保存)を目的として凍結保存を行っています。
更に、地震や火災などで貴重な動物が全滅した場合、凍結保存しておけば、個体を復元することができます。
当センターでは、遺伝子改変マウスが最も多く実験に使用されています。主に、疾患モデルとして、疾患の発症機序の解明や治療法の開発などに使用されています。これらの遺伝子改変マウスの作製も、当センターで行われています。特に、最近は、ゲノム編集(CRISPR/Cas9 system)によるノックアウトマウス、ノックインマウス、コンディショナルノックアウトマウス及び点変異マウス等を作製しています。
1.微生物クリーニング(清浄化)
他大学等から遺伝子改変マウスを導入する際、必ず微生物クリーニングを行います。
まず、遺伝子改変マウスの♂2匹に対して♀5~10匹用意します。あるいは、市販の♀マウスを使う場合もあります。♂からは精子を、♀からは卵子を採取し、体外受精用培地を用いて体外受精を行い、受精卵を得ます。これらの受精卵を1日培養して、2-細胞期胚に発生したものを偽妊娠0.5日目(プラグ確認日を0.5日とする)のレシピエントマウスの卵管内に移植します。移植後、19.5日目出産し、1ヵ月後に離乳しますので、離乳後にレシピエントマウスの微生物検査を行い、問題がなければ、他の飼育室へ移します。なお、出産数が少ないことが考えられる場合は、出産しても食殺及び育児放棄が生じるので、レシピエントマウスをsacrificeし、帝王切開にて胎児を取り出し、foster motherにnursingさせます。
2.凍結保存
体外受精由来2-細胞期胚をガラス化保存液(DPS; 2.75M DMSO, 2.75M propylene glycol, 1M sucrose)に浸漬し、直ちに液体窒素内にplungingすることにより行われます。DPSのようにDMSOなどの高濃度に含む溶液は、急速に冷却すると、氷点がなくなり、-196℃でも氷の結晶ができない状態で固化(solidification)します。この現象をガラス化(vitrification)といいます。ガラス化状態では、氷の結晶ができないので、白濁せず、ガラスのように透明になります。現在、マウス胚の保存は、ガラス化保存が主流になっています。
マウス精子の凍結保存は、精巣上体尾部精子を保存液(18% raffinose + 1.75% glycerol in M2 medium)中に懸濁させた後、クライオチューブかストローに移し、凍結保存容器 BICELLにチューブやストローを入れ、一晩、-80℃のディープフリーザー中で凍結させます。その後、液体窒素保存タンクに移して、保存します。
3.個体復元
凍結保存2-細胞期胚や精子を融解し、移植あるいは体外受精/移植により産仔を得ます。他大学や研究機関で凍結保存した胚や精子でも、融解方法がわかれば、個体化することができます。特に、海外からの遺伝子改変マウス等の輸入は、生体での輸送では、感染防御や動物愛護の観点から問題がありますので、凍結胚や凍結精子で輸送するケースが増加しています。
4.トランスジェニック(Tg) マウス作製
Tgマウスは、受精卵(1-細胞期卵)の精子由来前核(雄性前核)への外来DNAの顕微注入(pronuclear microinjection)かあるいは未受精卵への精子+外来DNAの顕微注入(cytoplasmic sperm injection; ICSI)のいずれかで行われています。当センターでは、主にICSIによってTgマウスを作製しています。
5.ノックアウト(Ko)マウス作製
ターゲット遺伝子に対するtargeting vector (例えば、pgk-neorとpgk-DT-Aを含む)を作製し、ES細胞にelectroporationによりtargeting vectorを導入します。導入後、相同組換えによりtargeting vectorを挿入することができます。その後、G418を含むES培養液にてES細胞を培養し、生存している細胞をピックアップし、クローニングを行います。これらのクローンについてsequencingを行い、targeting vectorが目的のサイトに挿入されているESクローンを選別します。これらのESクローンを別の系統(例えばICR、BALB/c等)の胚盤胞内に顕微注入します。顕微注入後、レシピエントマウスの子宮角内に移植し、キメラマウスを作製します。これらのキメラマウスと野生型の♀マウスと交配させ、ヘテロマウスを得ます。また、ヘテロマウス同士の交配により、両アリルにtargeting vectorが組み込まれているホモのKoマウスを得ることができます。
6.ゲノム編集(CRISPR/Cas9)マウス作製
1) ノックアウト(Ko) マウス
ノックアウトしたいターゲット遺伝子に対するsgRNAを合成し、Cas9蛋白質あるいはmRNAと共に受精卵(1-細胞期卵)の雄性前核かあるいは細胞質に顕微注入します。注入後、一晩、培養し、2-細胞期胚に発生したものを偽妊娠0.5日目のレシピエントマウスの卵管内に移植します。移植19日目に出産しますが、生後1ヵ月目に離乳させ、産仔の尻尾より抽出したDNAを用いてT7E1 assayとsequencingを行い、ノックアウト(indel挿入)マウスを同定します。Sequencingの結果から、sgRNA/Cas9を顕微注入した受精卵由来の産仔は、ほぼ100% indel挿入が認められます。ES細胞を用いないので、かなり高率良くKoマウスを作製することができます。
2) ノックイン (KI) マウス
ノックインしたいドナーベクター、sgRNA及びCas9蛋白質(mRNA)を同時に受精卵(1-細胞期卵)の前核、細胞質あるいはICSIで顕微注入した後、2-細胞期胚に発生したものを偽妊娠0.5日目のレシピエントマウスの卵管内に移植します。出産した個体は、離乳後、尻尾よりDNAを抽出し、PCRによるgenotypingを行います。最終的には、PCR productsを用いてsequencingを行い、挿入されたドナーベクターを同定します。
セーフハーバー領域を有するRosa26 locusにプロモーター付きのcDNAをノックインすることで、安定して発現するノックインマウスを作製することもできます。
3) コンディショナルノックアウトマウス
Cre/loxP systemを適用したコンディショナルノックアウトマウスをゲノム編集で作製することができます。組換えにより除去したいターゲット遺伝子のexonの両端に存在するintronにloxP配列を各々ノックインにより挿入することができます。まず、片側のloxP配列を含むドナーベクター、sgRNA及びCas9蛋白質(mRNA)を受精卵(1-細胞期卵)の前核、細胞質あるいはICSIで顕微注入した後、培養し、2-細胞期胚に発生した時点で、2つの割球の細胞質に各々もう一方のloxP配列を含むドナーベクター、sgRNA及びCas9蛋白質(mRNA)を顕微注入します。注入後、直ちに偽妊娠0.5日目のレシピエントマウスの卵管内に移植します。出産した個体は、離乳後、尻尾よりDNAを抽出し、PCRによるgenotypingを行います。あるいは、一旦、片側のloxPを挿入されたノックインマウスを作製しておき、そのノックインマウスの受精卵(1-細胞期卵)に、もう一方loxP配列を含むドナーベクター、sgRNA及びCas9蛋白質(mRNA)を顕微注入し、両方のloxP配列をもつノックインマウスを作製することもできます。
4) 1塩基置換変異マウス
1塩基置換変異マウスは、1塩基変異を有するssODN、sgRNA及びCas9蛋白質(mRNA)を受精卵(1-細胞期卵)の前核、細胞質あるいはICSIで顕微注入した受精卵を卵管移植することにより得ることができます。
5) 大規模欠失ノックアウトマウス
欠失させたいexonの両側のintronにsgRNAを各々設定し、2種のsgRNA、Cas9蛋白質(mRNA)を同時に受精卵(1-細胞期卵)の前核あるいは細胞質に顕微注入することにより、各々のsgRNAにおけるtarget 配列を同時に切断(indel mutation)することができます。即ち、exonを欠失させたノックアウトマウスを作製することができます。
※下記は全て学内限定
・微生物クリーニング依頼書
・受精卵配偶子凍結保存依頼書
・受精卵配偶子融解依頼書
・トランスジェニック(ゲノム編集)動物作製依頼書
7.アデノウイルスベクター作製
(問い合わせ先:中西友子 nakanishi-t@juntendo.ac.jp)
現在汎用されているアデノウイルスベクターは、非増殖型と呼ばれています。もとになるウイルスであるヒトアデノウイルス5型は36 kbの直鎖状2本鎖DNAをゲノムとして持ち、小児にかぜを起こすウイルスの1つです。非増殖型アデノウイルスベクターでは、このウイルスの遺伝子発現・増殖に必須なE1AとE1Bという2つの遺伝子を欠失させ、代わりに目的遺伝子の発現ユニットを組み込む構造となっています(研究室で開発されたアデノウイルスベクターは、タカラのAdenovirus dual expression kitとしてキット化されており、理研バイオリソースバンクhttps://dna.brc.riken.jp/ja/rvdja/adeno2jaにもpAxcwit2等として寄託済です)。E1A,E1B,E3を欠失した一般的なベクターでは、6.5 kbまでの大きな発現ユニットが組み込むことが可能です。血球系細胞への導入効率は低いものの、げっ歯類を含む動物細胞、鳥類の細胞など多くの細胞種に高い遺伝子導入効率を示し、神経など静止期の細胞への遺伝子導入も可能です。きわめてウイルス力価が高い10^9プラーク形成単位/mlのウイルス液が得ることができ、簡便な濃縮法による高濃度ベクター調製も容易です。一方、アデノウイルスベクターは、AAVベクターやレンチベクターと比較して搭載DNA量が大きいという長所を持ちますが、細胞染色体上への積極的な組込み機構がないので、細胞あたり20コピー導入されたとしても数回の細胞分裂で平均1コピーを割ってしまう短所もあります。しかし見方を変えると「トランスフェクションの効率が100%になったもの」とも言えます。このような優れた性質を持つことから、アデノウイルスベクターは基礎研究や遺伝子治療など広範な研究に有用性を発揮しています。我々は、E1領域にEFプロモーターを外向きに搭載することで、免疫反応を誘起するAdV由来pIXの発現や免疫反応の問題は回避できることも報告しています(Hum Gene Ther. 2007, 18:925-36; Virus Res. 2011.160:89-97)。
コスミドベクターの構築からアデノベクターの作製の流れについてはこちらをご参照ください。
8.ゲノム編集用アデノウイルスベクター作製
(問い合わせ先:中西友子 nakanishi-t@juntendo.ac.jp)
アデノウイルスベクターの搭載可能DNAが大きいことを活かして、ガイドRNAを8個とspCas9の変異体であるCas9 nickaseを同時発現する一体型のアデノウイルスベクターの開発に成功し、ダブルニッキング切断により高効率で安全にゲノム編集可能なシステムの構築しています(論文3件、特許2件、日経産業新聞 2019年8月14日)。次世代シークエンスによる解析により、Cas9 nickaseではCas9を使用した場合と比較して、実際にオフターゲット変異が1000分の1以下(検出限界以下)であることを確認しています(図)。
現在、オートファジー関連遺伝子を培養細胞で多重にノックアウトできるベクターや、ヒトパピローマウイルスやヒトB型肝炎ウイルスのゲノムを破壊することで子宮頸がんや肝がんのゲノム編集治療を目指すベクターを作製し共同研究を進めています。また、ドナーDNAもベクターに搭載することで先天性代謝異常症の一つであるフェニルケトン尿症をノックインにより治療可能なモデル系の開発も試みています。
Cas9/Cas9 nickase発現アデノベクターは、理研バイオリソースバンクに寄託済(https://dna.brc.riken.jp/ja/gene_analysis/crispr_cas9)です。 Tetraplex‑guide Tandem 法(Sci Rep. 2021, 11: 3961)を利用したコスミドベクターの構築およびアデノベクター作製の流れについてはこちらをご参照ください。
1. Kato Y, Saito I, *Nakanishi T (Last author), et al. Int J Mol Sci. 2021, 22(19):10570
2. Nakanishi T (Corresponding author), Saito I, et al. Sci Rep. 2021, 11(1): 3961
3. Nakanishi T, Saito I, et al. J Gene Med. 2019, 21(11): e3115
4. 中西友子、斎藤泉、清野透、中原知美、出願番号:特願2023-014899
5. 中西友子、斎藤泉、出願番号:PCT/JP2019/037255